Insulinresistens – det nya paradigmet
Här följer ett översatt gästinlägg av dr Jason Fung, kanadensisk njurspecialist och världsledande expert på periodisk fasta och LCHF:
Det paradigm vi i nuläget tror på gällande insulinresistens är nyckel- och nyckelhålsmodellen, och det är helt enkelt fel.
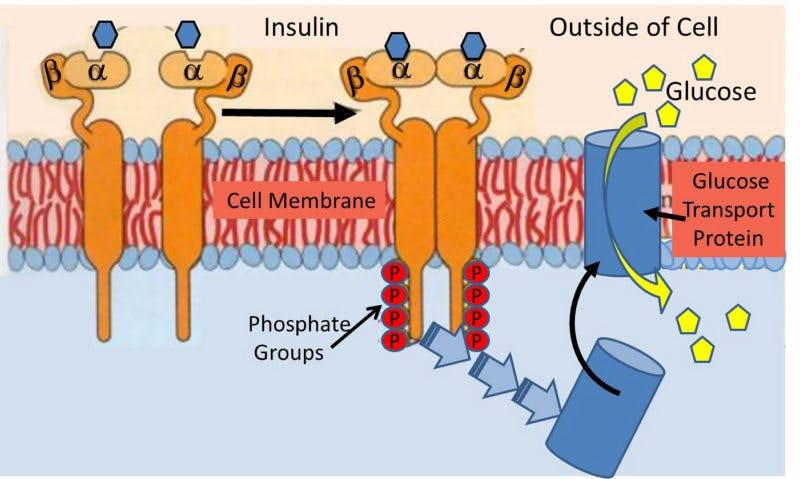
Insulin är ett hormon som verkar på en hormonreceptor på cellmembranet för att få sin effekt. Detta kallas ofta nyckel- och nyckelhålsmodellen.
Nyckelhålet är insulinreceptorn som ser till att dörrarna in till cellen är stängda. När rätt nyckel (insulin) sätts in öppnas dörrarna för att låta glukos från blodet in i cellen. Detta glukos används därefter som energi till cellen.
Tar man bort nyckeln (insulinet) stängs dörrarna och blodglukos kan inte längre komma in i cellen.
Nyckel- och nyckelhålsmodellen vid insulinresistens
Vad sker vid fenomenet med insulinresistens? Klassiskt sett föreställer vi oss att nyckeln och nyckelhålet inte längre passar. Nyckeln (insulinet) kan öppna nyckelhålet (receptorn) men bara delvis och inte särskilt bra. Resultatet blir att glukos inte kan passera genom dörren.
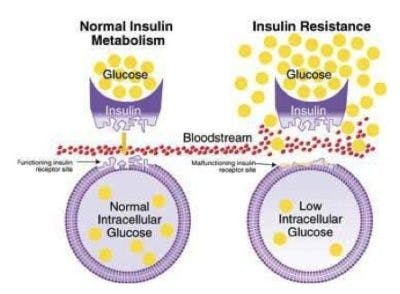 Detta resulterar i lägre nivåer av glukos i cellen än vad som anses vara normalt. Glukos, som nu blockeras av den stängda dörren, ackumuleras utanför cellen i blodet, vilket vi kan se genom förhöjt blodsocker då vi diagnosticerar någon med typ 2-diabetes.
Detta resulterar i lägre nivåer av glukos i cellen än vad som anses vara normalt. Glukos, som nu blockeras av den stängda dörren, ackumuleras utanför cellen i blodet, vilket vi kan se genom förhöjt blodsocker då vi diagnosticerar någon med typ 2-diabetes.
Detta har också beskrivits som ett internt svältläge eftersom cellen har väldigt lite socker på insidan. Kroppens reflexartade reaktion blir att öka insulinproduktionen (nyckeln). Eftersom varje nyckel fungerar sämre än den tidigare, så överproduceras antalet nycklar för att se till att glukos går in till cellerna. En bra och enkel teori.
Problemen
Problemet är att paradigmen inte stämmer överens med verkligheten. Först och främst, är problemet insulinet eller insulinreceptorn? Det är väldigt lätt att ta sig en titt på insulinets struktur och insulinreceptorns struktur i insulinresistenta patienter. Det enda som krävs är att isolera insulinet eller några celler och sedan kolla på deras struktur med avancerade molekylärverktyg. Då blir det genast uppenbart att det varken är fel på insulinet eller receptorn. Så vad är då problemet?
Den enda återstående möjligheten är att det är någonting som klistrar igen systemet. Någon typ av blockering som kommer emellan nyckel- och nyckelhålsmekanismerna. Men vad är det för något? Det finns alla möjliga sorters teorier. Inflammation. Oxidativ stress. Advanced glycation end products (AGEs). Alla vanliga slagord som doktorer slänger med när de egentligen inte har någon aning om vad som pågår. Med denna modell går det inte att förklara vad som orsakade insulinresistens. Utan förståelse för vad som orsakar insulinresistens har vi inte någon chans att behandla det.
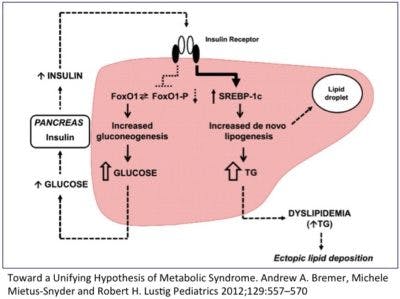 Så finns den centrala paradoxen av insulinresistens i levern. Låt mig förklara. Insulin kan påverka levern på två sätt. Kom ihåg att insulinnivåerna stiger då du äter. Det säger åt kroppen att sluta producera glukos i levern (glukoneogenesen) eftersom det finns en massa glukos som kommer in från magen (mat). Detta styrs genom FOX01-vägen.
Så finns den centrala paradoxen av insulinresistens i levern. Låt mig förklara. Insulin kan påverka levern på två sätt. Kom ihåg att insulinnivåerna stiger då du äter. Det säger åt kroppen att sluta producera glukos i levern (glukoneogenesen) eftersom det finns en massa glukos som kommer in från magen (mat). Detta styrs genom FOX01-vägen.
Den andra stora påverkan som insulin har på levern är att det ökar fettproduktionen (De novo-lipogenesen (DNL)). Detta för att hantera den inkommande strömmen av glukos som kroppen inte kan ta hand om direkt. Detta styrs genom SREBP-1c-vägen.
Så om levern blir insulinresistent avtar insulinets effekt på båda dessa vägar. Med andra ord, levern fortsätter producera glukos, och slutar producera fett. Men det är bara sant i fallet med glukoneogenesen. Vid insulinresistens fortsätter levern producera nytt glukos precis som man kan förvänta sig. Men DNL (produktionen av nytt fett) fortsätter och ökar faktiskt. Så insulinets effekt på DNL är inte dämpat utan accelererande!
Vad?
Hur kan det ens vara möjligt att denna insulinresistenta lever selektivt är insulinresistent för en effekt av insulin på levern men sedan samtidigt accelerera den andra verkan? I samma cell, som svar på samma mängd insulin, med exakt samma insulinreceptor? Det verkar ju helt galet. Samma cell är alltså insulinresistent och insulinkänslig på samma gång!
En bättre förklaring: översvämning
Hur kan man förklara denna paradox?
Det behövs ett paradigmskifte för insulinresistens som passar verkligheten bättre. Faktum är att vi kan se på insulinresistens som ett översvämningsfenomen, istället för att använda nyckel- och nyckelhålsmodellen. Allt vi vet om insulinresistens är att det är mycket svårare att få in glukos i en ”insulinresistent” cell än i en vanlig cell.
Men detta innebär inte nödvändigtvis att dörren är stängd. Istället kanske det är så att cellen håller på att översvämmas av glukos och det då inte går att trycka in mer.
Se på cellen som en tunnelbanevagn. När dörren öppnas stiger passagerarna på från utsidan (glukos i blodet) in till den tomma tunnelbanevagnen (cellen). Normalt sett krävs det inte mycket för att få glukos in i cellen (insulinet trycker in det).
Men vid insulinresistens är inte problemet att cellen inte öppnar sig. Problemet är istället att tunnelbanevagnen (cellen) redan håller på att översvämmas av passagerare (glukos). Nu kan glukos utanför cellen helt enkelt inte ta sig in och det blir istället överfyllt på perrongen.
 Insulin försöker trycka in glukos i cellen precis som de som jobbar med att trycka in folk i tunnelbanevagnar i Japan, men det går helt enkelt inte eftersom cellen redan är full. Så den reflexartade reaktionen blir att producera mer insulin (intryckare) för att få glukos att komma in i cellen. Vilket fungerar, men bara under kort tid.
Insulin försöker trycka in glukos i cellen precis som de som jobbar med att trycka in folk i tunnelbanevagnar i Japan, men det går helt enkelt inte eftersom cellen redan är full. Så den reflexartade reaktionen blir att producera mer insulin (intryckare) för att få glukos att komma in i cellen. Vilket fungerar, men bara under kort tid.
Så cellen befinner sig inte i svältläge. Istället blir det översvämning av glukos som börjar läcka ut i blodet. Detta ser ut som glukoneogenes men det är inte riktigt förenligt med insulinresistens. Men vad händer med fettproduktion?
I den klassiska insulinresistensmodellen är paradoxen att DNL ökar, och inte minskar, vilket ser ut som ökad insulinkänslighet istället för resistens. Men i översvämningsmodellen ökar DNL för att cellen försöker bli av med överflödigt glukos genom att producera extra fett. Cellen översvämmas alltså och är inte i svältläge.
Varför är det viktigt?
Varför är detta väldigt viktigt? Eftersom förståelse för denna nya paradigm kommer att leda till ett svar på frågan hur insulinresistens utvecklas och vad vi kan göra mot det. Problemet är varken insulin eller insulinreceptorn. Båda är normala. Problemet är att cellen är proppfylld med glukos. Så vad har orsakat det?
Svaret verkar nu uppenbart – det rör sig om för mycket glukos och för mycket insulin. Med andra ord, insulinet var orsaken till att insulinresistens utvecklades. Vi behöver inte jaga skuggor för att söka efter den mystiska orsaken till insulinresistens.
Då vi börjar förstå varför för mycket glukos och insulin är roten till insulinresistens kan vi också ge patienter en rationell behandling. Minska insulin- och glukosnivåerna. När insulinresistensen reverseras, botas typ 2-diabetes.
Ett bättre sätt
Hur man reverserar typ 2-diabetes
Tidigare med dr Fung
Hur du fixar din trasiga metabolism genom att göra precis tvärtom
Misslyckandet för The Biggest Loser och succén med den ketogena studien
Videor


















Mer med dr Fung
Dr Fung har sin egen blogg på intensivedietarymanagement.com. Han är också aktiv på Twitter.
Jason Fungs bok The Obesity Code finns tillgänglig på Bokus.
